
Clear Reporting Responsibilities for U.S. Trials
Understand PI and site reporting obligations under U.S. regulations, including when an adverse event has to go to the sponsor, the IRB, or another oversight body, and when documenting it is enough.
Timelines, Triggers, and Decision Points
Learn what starts a reporting clock, how seriousness and relatedness change what's expected, and how to work through the gray areas without over- or under-reporting.
Built for Real-World Site Reporting
Focused on how reporting happens at a site, so your documentation stays consistent and your reporting calls hold up.
From Definitions to Defensible Reporting
Adverse event reporting is where definitions turn into action. It's also where site teams feel the most pressure to get it right, because this is the part regulators see. This session focuses on PI and site reporting responsibilities under U.S. regulations, and on how safety information moves from your site to sponsors, IRBs, and oversight bodies. You'll learn how reporting expectations change by event type, what triggers a timeline, and how to document and justify a reporting decision in real practice. We walk through how sites decide what needs reporting, who needs to know, and when, including sponsor notification, IRB reporting, and follow-up. We also clear up the spots that confuse people most, like events that are serious but expected, events that are possibly related, and the times documentation matters even when formal reporting doesn't. If you want confidence in your reporting decisions instead of guesswork, start here.
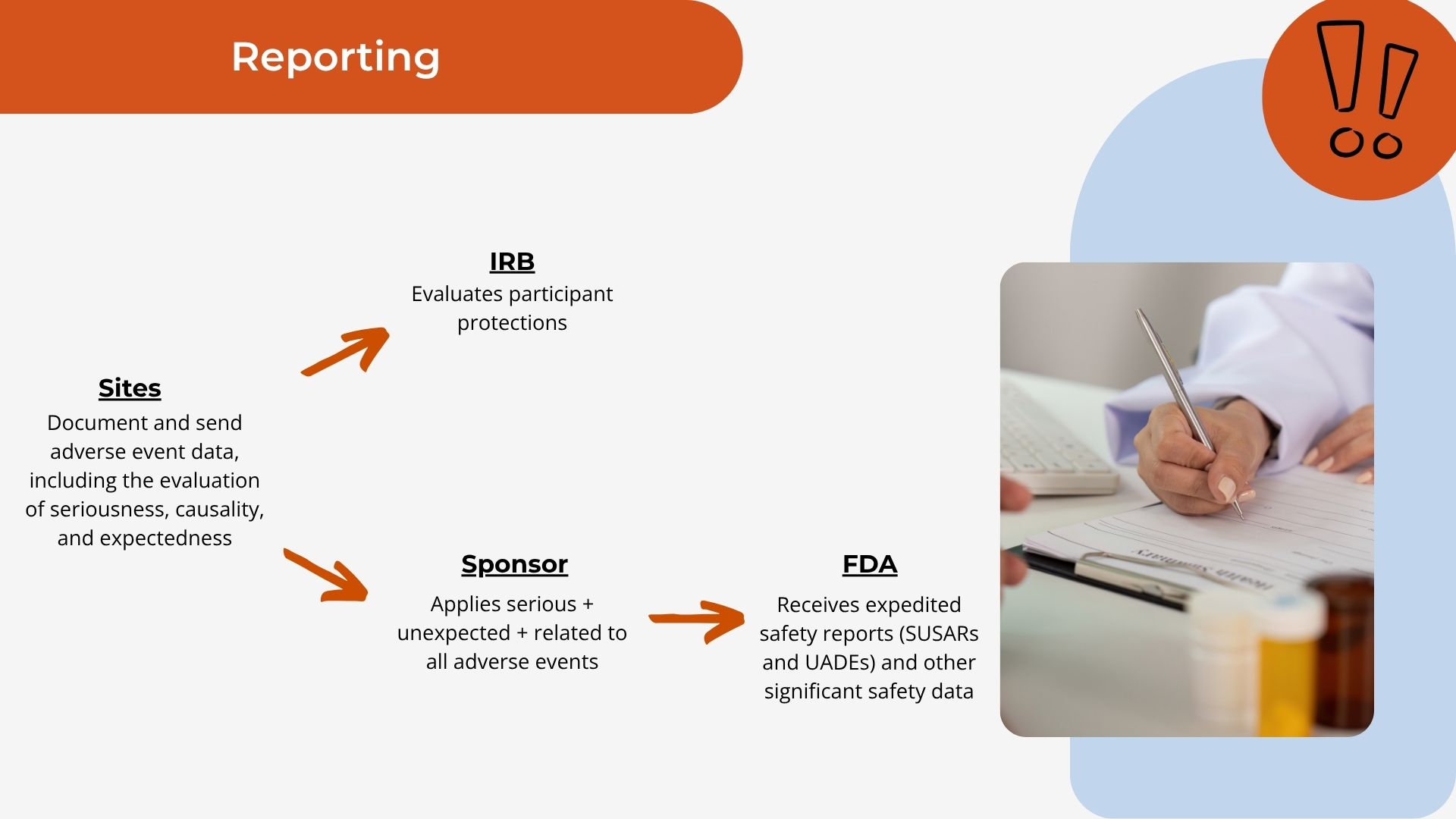
The reporting flow mapped.
Where safety information goes, from your site to the sponsor to the FDA.
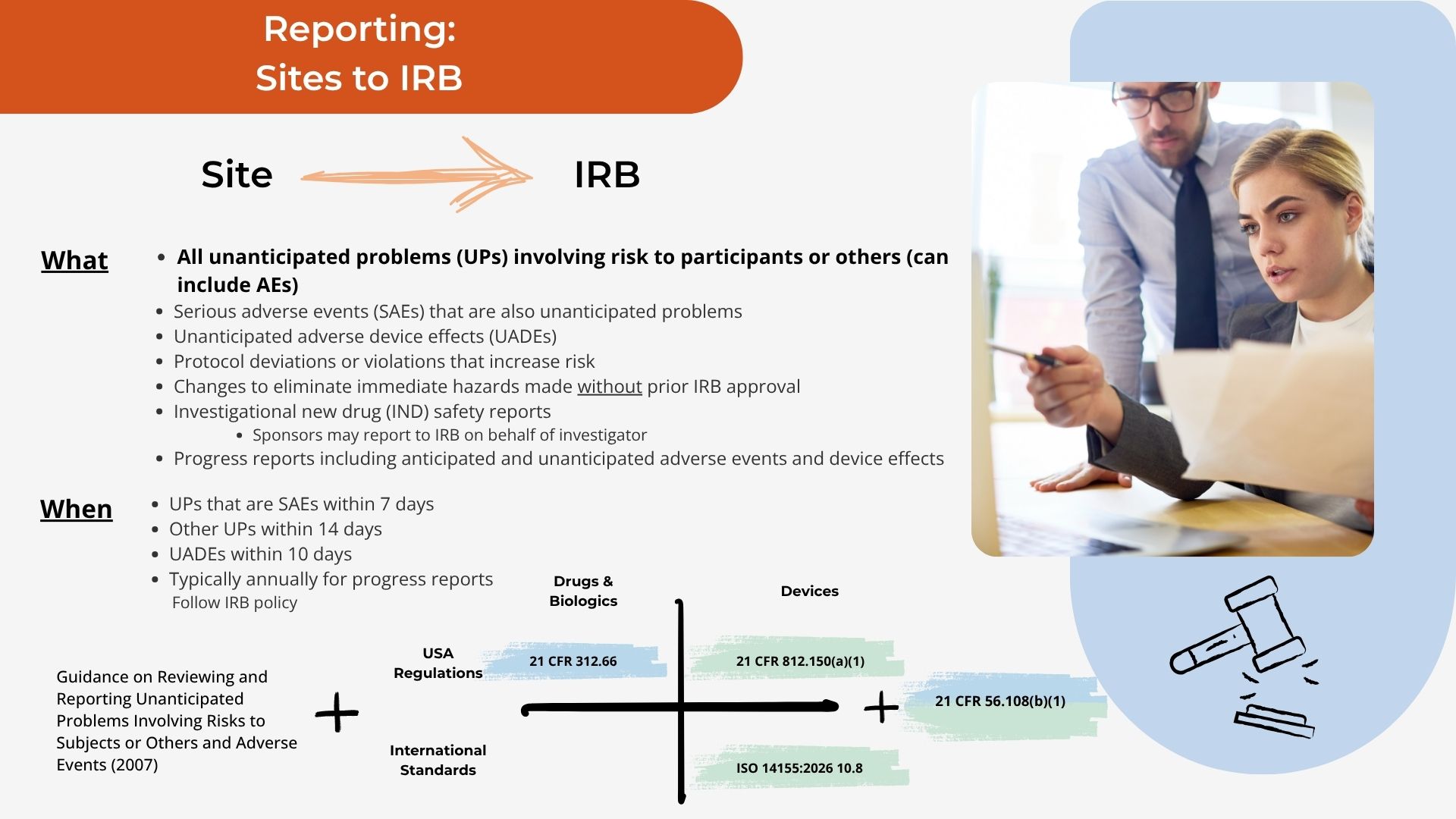
Grounded in the regulations.
Every requirement tied back to the CFR and standard section it comes from.
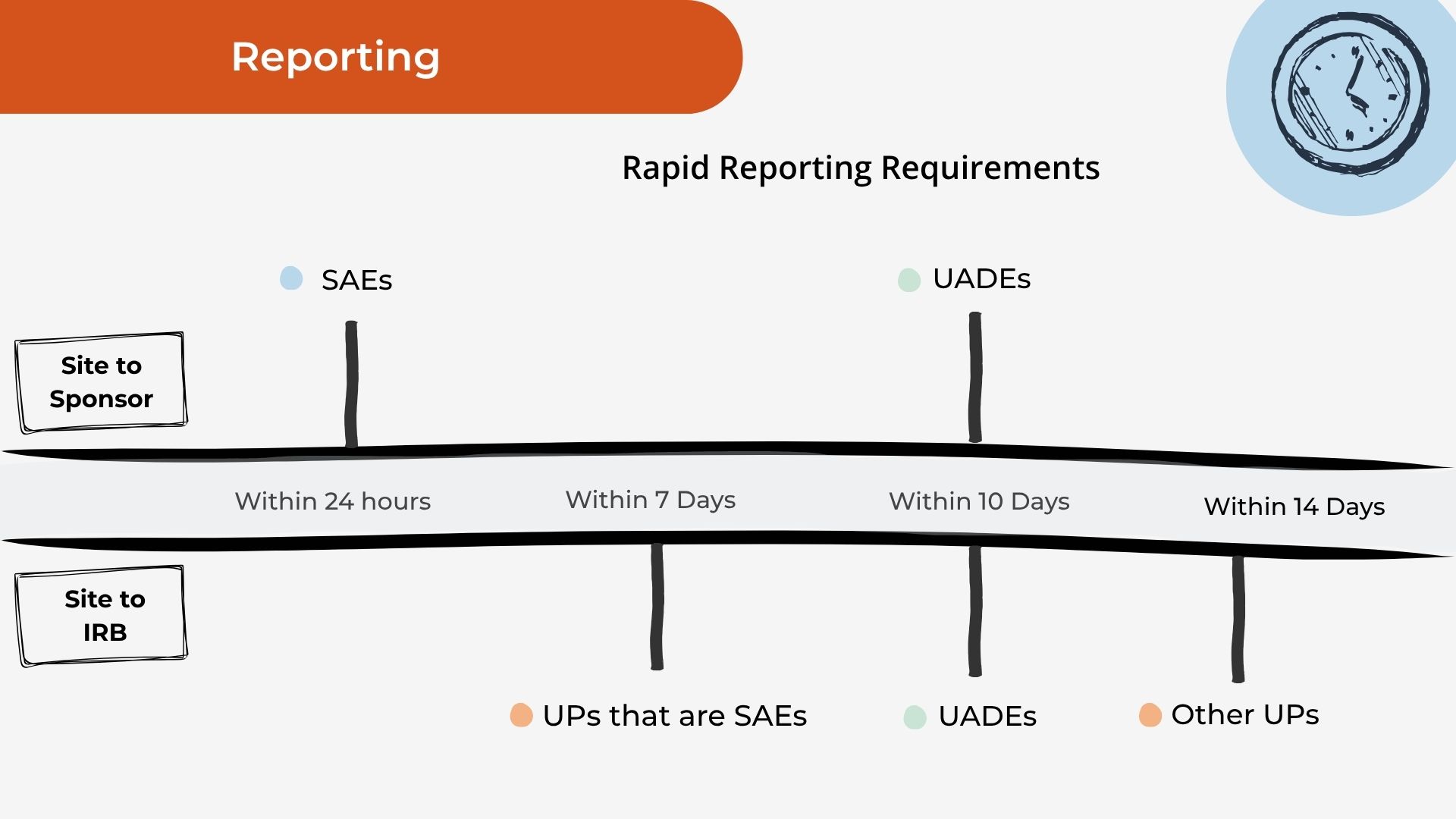
Know your deadlines.
The reporting clocks for sites, drawn out so none slips by.
Want to keep building your safety reporting foundation?

This session is part of the Safety Reporting for Sites: A Foundations Series bundle, which brings four safety reporting sessions together into one cohesive learning path. Available in the ClinIQ Academy catalog.
Curriculum
-
1
-
(Included in full purchase)
Safety Reporting for Sites: Site Reporting Requirements (Drugs & Devices)
-
(Included in full purchase)
Reporting that protects participants and your site.
Get clear on what's expected, and make every safety reporting call with confidence instead of a guess.
$10.00